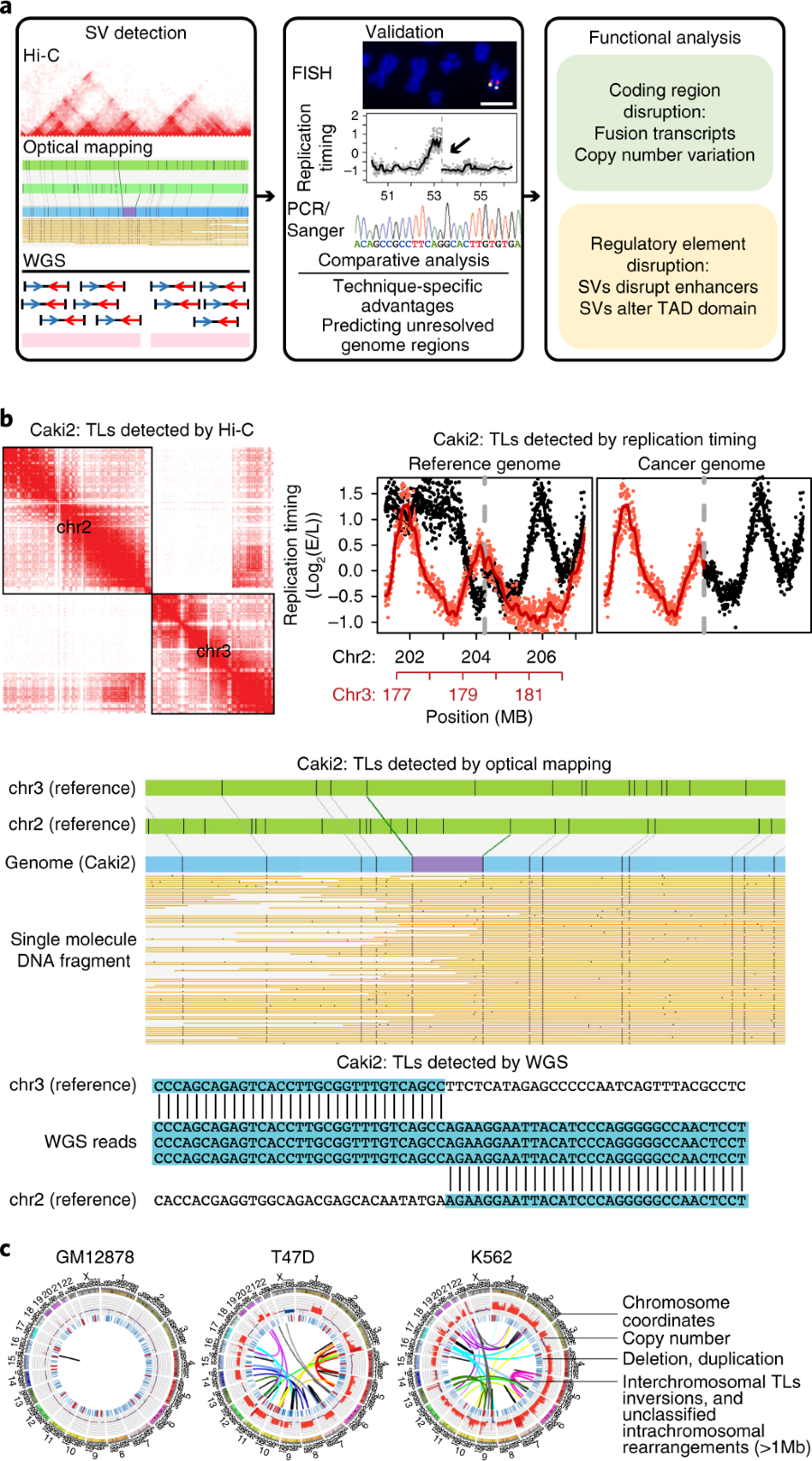
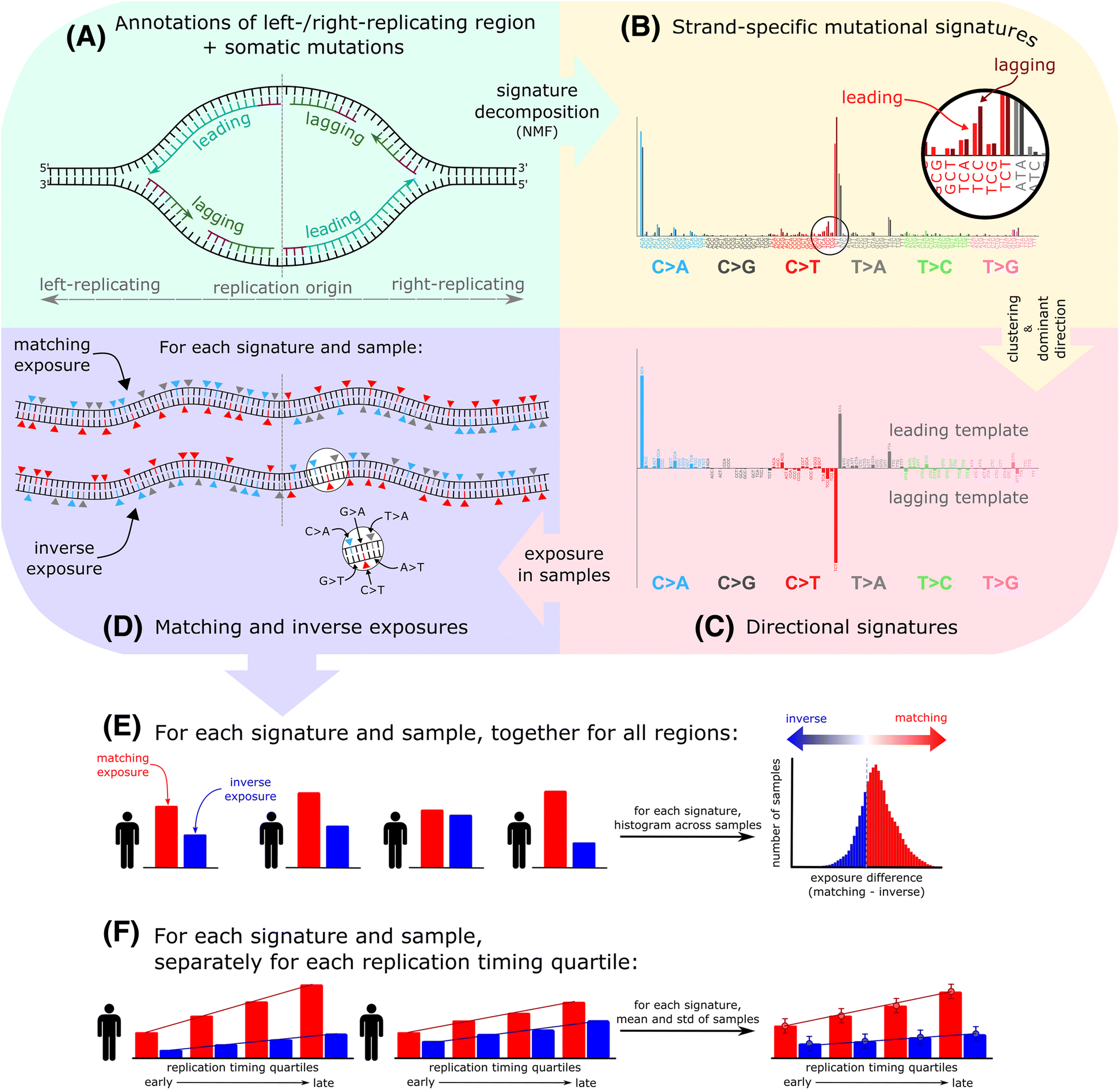
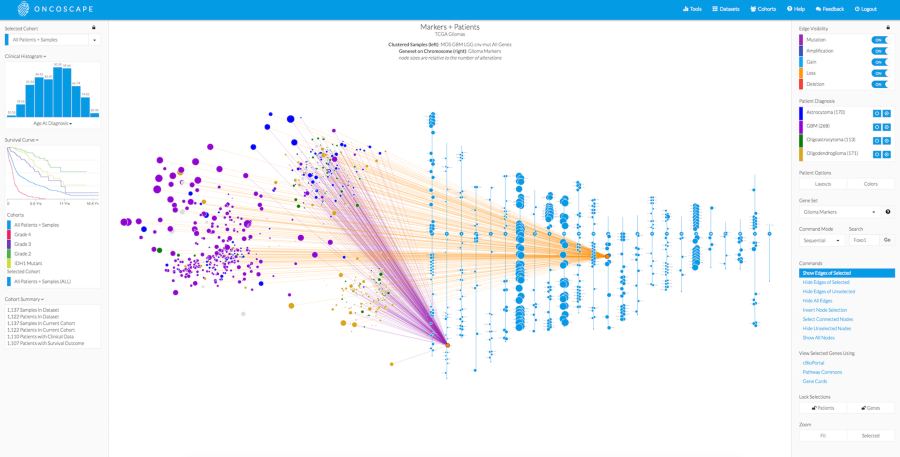
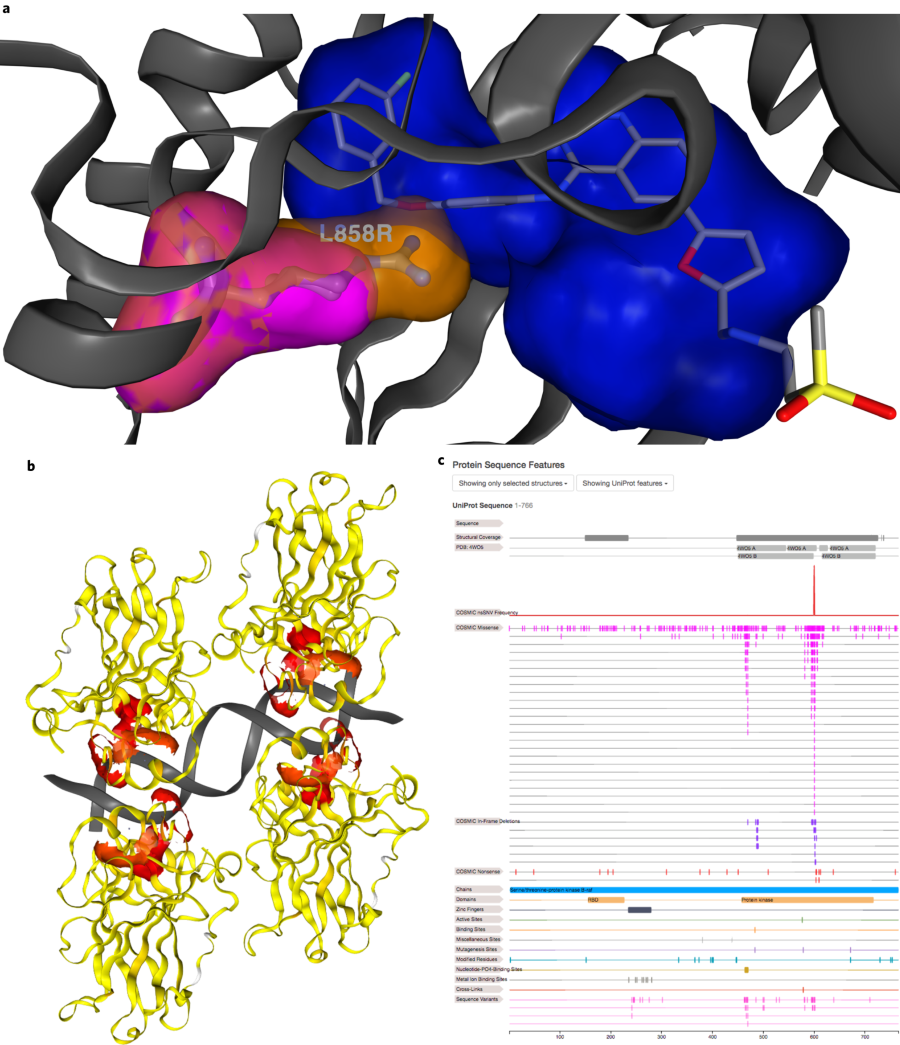
[] [Bioinformatics]
Pisces: An Accurate and Versatile Variant Caller for Somatic and Germline Next-Generation Sequencing Data

“a rapid, versatile and accurate small variant calling suite designed for somatic and germline amplicon sequencing applications. Accuracy is achieved by four distinct modules, each incorporating a number of novel algorithmic strategies.”